What is in this leaflet
This leaflet answers some common questions about ANDEPRA. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
The information in this leaflet was last updated on the date shown on the final page. More recent information on this medicine may be available. Make sure you speak to your pharmacist or doctor to obtain the most up to date information on this medicine. You can also download the most up to date leaflet from www.lilly.com.au. The updated leaflet may contain important information about ANDEPRA and its use that you should be aware of.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking ANDEPRA against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What ANDEPRA is used for
ANDEPRA is used to treat major depressive disorder (depression), Diabetic Peripheral Neuropathic Pain (nerve pain caused by diabetes) and Generalised Anxiety Disorder (excessive worry)
ANDEPRA belongs to a group of medicines called Serotonin and Noradrenaline Reuptake Inhibitors (SNRIs). SNRIs are believed to work by their action on serotonin and noradrenaline in the brain. Serotonin and noradrenaline are the chemical messengers responsible for controlling the psychological and painful physical symptoms of depression.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason. This medicine is available only with a doctor's prescription.
ANDEPRA is not recommended for use in children and adolescents under the age of 18 years.
Before you take ANDEPRA
When you must not take it
Do not take ANDEPRA if you have an allergy to:
- any medicine containing duloxetine hydrochloride;
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include shortness of breath, wheezing or difficulty breathing; swelling of the face, lips, tongue or other parts of the body; rash, itching or hives on the skin.
Do not take ANDEPRA if you have liver disease. This could increase the chance of you having liver problems during treatment with ANDEPRA.
Do not take this medicine if you are taking another medicine for depression called a monoamine oxidase inhibitor (MAOI) or have been taking a MAOI within the last 14 days. Check with your doctor or pharmacist if you are unsure as to whether or not you are taking a MAOI.
If you do take ANDEPRA while you are taking a MAOI, you may experience shaking (tremor), shivering, muscle stiffness, fever, rapid pulse, rapid breathing or confusion.
Do not take ANDEPRA if you are taking another medicine for depression called fluvoxamine.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any medical conditions, especially if you have:
- a condition in which the pressure of fluid in the eye may be high (glaucoma)
- high blood pressure
- heart problems
- kidney problems as you may need to take a lower dose of ANDEPRA
- history of fits (seizures)
- bipolar disorder
- diabetes
If you have high blood pressure or heart problems your doctor may monitor your blood pressure.
Tell your doctor if you are pregnant or plan to become pregnant or are breast-feeding. Your doctor can discuss with you the risks and benefits involved. If ANDEPRA is taken during pregnancy, you should be careful, particularly at the end of pregnancy. Transitory withdrawal symptoms have been reported rarely in the newborn after maternal use in the last 3 months of pregnancy.
Talk to your doctor about how much alcohol you drink. People who drink excessive amounts of alcohol should not take ANDEPRA. Drinking too much alcohol could increase the chance of you having liver problems during treatment with ANDEPRA.
If you have not told your doctor about any of the above, tell them before you start taking ANDEPRA.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and ANDEPRA may interfere with each other. These include:
- monoamine oxidase inhibitors (MAOIs), medicines used to treat some types of depression.
You should stop taking MAOIs at least two weeks before starting ANDEPRA.
You must stop taking ANDEPRA at least 5 days before you start taking a MAOI. - other medicines used to treat depression, panic disorder, anxiety or obsessive illnesses, including tryptophan
- strong painkillers such as tramadol, pethidine
- a type of migraine treatment called 'triptans', such as sumatriptan or zolmitriptan
- medicines used to treat stress urinary incontinence such as tolteridone
- medicines used to treat heart problems such as flecainide or propafenone
- thioridazine, a medicine used to treat schizophrenia
- herbal medicines such as St John's Wort (Hypericum perforatum)
- warfarin, a medicine used to thin the blood (anticoagulant) or other medicines known to affect blood coagulation (NSAIDs, aspirin)
Do not start to take any other medicine unless prescribed or approved by your doctor. These medicines may be affected by ANDEPRA or may affect how well it works. You may need different amounts of your medicines or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to take ANDEPRA
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the carton, ask your doctor or pharmacist for help.
How much to take
The usual recommended dose of ANDEPRA in Major Depressive Disorder or Diabetic Peripheral Neuropathic Pain is one 60 mg capsule taken once daily.
The recommended dose of ANDEPRA in Generalised Anxiety Disorder is between 30 mg and 120 mg, taken once daily.
Your doctor may start you on a lower dose to help reduce side effects.
If you have severe kidney disease, the recommended starting dose of ANDEPRA is one 30 mg capsule taken once daily.
How to take it
Swallow the capsule whole with a full glass of water.
Do not open the capsules and crush the pellets inside because the medicine may not work as well. ANDEPRA may be taken with or without meals.
When to take it
Take your medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
How long to take it
The length of treatment with ANDEPRA will depend on how quickly your symptoms improve. Most medicines of this type take time to work so don't be discouraged if you do not feel better right away.
Although you may notice an improvement, continue taking your medicine for as long as your doctor tells you.
If you forget to take it
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Otherwise, take it as soon as you remember, and then go back to taking your medicine as you would normally.
Do not take a double dose to make up for the dose that you missed.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much ANDEPRA. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
Symptoms of an overdose may include drowsiness, convulsions, and vomiting. Symptoms may also include some or all of the following: feeling confused, feeling restless, sweating, shaking, shivering, hallucinations, muscle jerks, fast heart beat.
While you are taking ANDEPRA
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking ANDEPRA.
Tell any other doctors, dentists and pharmacists who treat you that you are taking this medicine.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine
If you become pregnant while taking this medicine, tell your doctor immediately.
Keep all of your doctor's appointments so that your progress can be checked. Your doctor may do some tests from time to time to make sure the medicine is working and to prevent unwanted side effects.
Tell your doctor immediately if you have any suicidal thoughts or other mental/ mood changes. Occasionally, the symptoms of depression or other psychiatric conditions may include thoughts of harming yourself or committing suicide. These symptoms may continue or get worse during the first one or two months of treatment, until the full antidepressant effect of the medicine becomes apparent This is more likely to occur in young adults under 25 years of age.
Contact your doctor or a mental health professional right away or go to the nearest hospital for treatment if you or someone you know is showing any of the following warning signs of suicide:
- worsening of your depression
- thoughts or talk of death or suicide
- thoughts or talk of self-harm or harm to others
- any recent attempts of self-harm
- increase in aggressive behaviour, irritability or any other unusual changes in behaviour or mood.
All mentions of suicide or violence must be taken seriously.
If you notice any of the following contact your doctor right away.
Your doctor may do some blood tests to check your liver or tell you to stop taking your medicine. Signs of liver problems include:
- itchy skin
- dark urine
- yellowing of the skin or eyes
- tenderness over the liver
- symptoms of the 'flu'
These may be signs of serious liver damage.
Things you must not do
Do not take ANDEPRA to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking ANDEPRA or lower the dosage without checking with your doctor. If you stop taking it suddenly, your condition may worsen or you may have unwanted side effects. If possible, your doctor will gradually reduce the amount you take each day before stopping the medicine completely.
Do not drive or operate machinery until you know how ANDEPRA affects you. ANDEPRA may cause dizziness or drowsiness in some people.
Do not let yourself run out of ANDEPRA over the weekend or on holidays.
Things to be careful of
Be careful when drinking alcohol while you are taking this medicine. Drinking large amounts of alcohol during treatment with ANDEPRA can cause severe liver injury.
You should avoid 'binge drinking' or drinking excessively during treatment with ANDEPRA. Drinking alcohol with this medicine may also cause dizziness or drowsiness in some people. If you have any of these symptoms, do not drive, operate machinery or do anything else that could be dangerous.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking ANDEPRA. This medicine helps many people with depression, but it may have unwanted side effects in a few people. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have. The following side effects are the more common side effects of ANDEPRA and are often mild and short-lived.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- dry mouth, mouth ulcers, thirst, bad taste
- burping or belching, indigestion, stomach pain, nausea, vomiting,
- constipation, diarrhoea, wind (flatulence)
- bad breath
- loss of appetite, weight loss
- headache
- trouble sleeping
- dream abnormalities
- drowsiness
- feeling tired or having no energy
- sexual problems
- dizziness
- tremor
- blurred vision
- feeling anxious, agitated or restless
- confusion and attention problems
- tingling and numbness of hands, face, mouth and feet
- yawning or throat tightness
- difficulty urinating (passing water), urinating frequently or needing to urinate at night
- irregular heart beat
- hot and cold sweats
- sore ears, sore throat
- ringing in ears
- muscle pain, stiffness or twitching
- walking problems
- flushing
- skin rash
- restless legs
These are the more common side effects of your medicine.
Tell your doctor immediately if you notice any of the following:
-
signs of a possible serious liver problem,
such as nausea, vomiting, loss of appetite, feeling generally unwell, fever, itching, yellowing of the skin and/or eyes, dark urine - high pressure in the eye (glaucoma)
- feeling tired, weak or confused and having achy, stiff or uncoordinated muscles.
This may be because you have low sodium levels in the blood (hyponatraemia or syndrome of inappropriate antidiuretic hormone) - abdominal pain, traces of blood in your stools, or if your stools are dark in colour.
- This may because you have increased bleeding, possibly in the gastric tract (gastrointestinal bleeding). You may also feel weakness, dizziness and experience nausea and/or vomitting
- seeing or hearing things (hallucinations)
- dizziness or fainting when you stand up, especially from a lying or sitting position
- uncontrollable movements
- if you have some or all of the following symptoms you may have something called serotonin syndrome: feeling confused, feeling restless, sweating, shaking, shivering, hallucinations, sudden jerks in your muscles or a fast heart beat
- stiff neck or jaw muscles (lockjaw)
- fits or seizures
- mood of excitement, over-activity and uninhibited behaviour.
- aggression or anger especially after starting or stopping taking this medicine
You may need urgent medical attention.
Other changes you may not be aware of:
- increased blood pressure
- heart rhythm changes
- underactive thyroid gland
- liver function changes
If any of the following happen, tell your doctor immediately or go to Accident and Emergency at your nearest hospital:
- itching, skin rash or hives
- shortness of breath, wheezing or trouble breathing
- swelling of the face, lips, tongue or other parts of the body
These are very serious side effects. You may need urgent medical attention or hospitalisation.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell. This is not a complete list of all possible side effects. Others may occur in some people and there may be some side effects not yet known.
After taking ANDEPRA
Storage
Keep your capsules in the pack until it is time to take them. If you take the capsules out of the pack they may not keep as well.
Keep your capsules in a cool dry place where the temperature stays below 25°C.
Do not store ANDEPRA or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
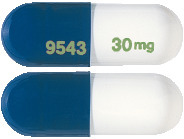
- ANDEPRA 30 mg capsules have an opaque white body and opaque blue cap imprinted with "9543" and "30 mg" using green ink and are available in packs of 7(starter packs) and 28.
- ANDEPRA 60 mg capsules have an opaque green body and opaque blue cap imprinted with "9542" and "60 mg" using white ink and are available in packs of 7(starter packs) and 28.
Ingredients
Active Ingredients
- 30 mg capsule - duloxetine hydrochloride equivalent to duloxetine 30 mg
- 60 mg capsule - duloxetine hydrochloride equivalent to duloxetine 60 mg
Inactive Ingredients
- indigo carmine CI73015
- gelatin
- hypromellose
- hypromellose acetate succinate
- sodium lauryl sulfate
- sucrose
- sugar spheres
- talc - purified
- titanium dioxide
- triethyl citrate
- iron oxide yellow CI77492 (60mg capsules)
- TekPrint SB-4028 Green Ink (30mg capsules)
- TekPrint SB-0007P White Ink (60mg capsules)
This medicine does not contain lactose, gluten, tartrazine or any other azo dyes.
Suppliers
Eli Lilly Australia Pty Limited
112 Wharf Road
WEST RYDE, NSW 2114
®= Registered Trademark
Australian Registration Number
- 30 mg capsules: AUST R 179186
- 60 mg capsules: AUST R 179187
This leaflet was prepared in March 2012.
Published by MIMS April 2013

 The following additional adverse events were reported during placebo controlled clinical trials of duloxetine for MDD or other indications in 8504 patients. Very common events are defined as those occurring in ≥ 10% of patients, common events are defined as those occurring in ≥ 1% and < 10% of patients, uncommon events are defined as those occurring in ≥ 0.1% and < 1% of patients, and rare events are defined as those occurring in < 0.1% of patients.
The following additional adverse events were reported during placebo controlled clinical trials of duloxetine for MDD or other indications in 8504 patients. Very common events are defined as those occurring in ≥ 10% of patients, common events are defined as those occurring in ≥ 1% and < 10% of patients, uncommon events are defined as those occurring in ≥ 0.1% and < 1% of patients, and rare events are defined as those occurring in < 0.1% of patients. In addition to the HAMD17 total score, several other measures were included in the evaluation of efficacy of duloxetine. HAMD17 Depressed Mood Item (Item 1), the Anxiety Subfactor of the HAMD17, the Patient Global Impressions (PGI) Improvement Score, bodily pain as measured by Visual Analog Scale (VAS), and the Quality of Life in Depression rating scales were also examined. In the four studies where duloxetine demonstrated statistical superiority over placebo as measured by improvement in the HAMD17 total score, results were also positive for the additional measures at doses of 60 mg to 120 mg per day.
In addition to the HAMD17 total score, several other measures were included in the evaluation of efficacy of duloxetine. HAMD17 Depressed Mood Item (Item 1), the Anxiety Subfactor of the HAMD17, the Patient Global Impressions (PGI) Improvement Score, bodily pain as measured by Visual Analog Scale (VAS), and the Quality of Life in Depression rating scales were also examined. In the four studies where duloxetine demonstrated statistical superiority over placebo as measured by improvement in the HAMD17 total score, results were also positive for the additional measures at doses of 60 mg to 120 mg per day. Patients enrolled had Type I or II diabetes mellitus with a diagnosis of painful distal symmetrical sensorimotor polyneuropathy for at least 6 months. The patients had a baseline pain score of ≥ 4 on an 11 point scale ranging from 0 (no pain) to 10 (worst possible pain). Patients were permitted up to 4 g of paracetamol per day as needed for pain, in addition to Andepra. Patients recorded their pain daily in a diary.
Patients enrolled had Type I or II diabetes mellitus with a diagnosis of painful distal symmetrical sensorimotor polyneuropathy for at least 6 months. The patients had a baseline pain score of ≥ 4 on an 11 point scale ranging from 0 (no pain) to 10 (worst possible pain). Patients were permitted up to 4 g of paracetamol per day as needed for pain, in addition to Andepra. Patients recorded their pain daily in a diary. For various degrees of improvement in pain from baseline to study endpoint, Figure 1 and Figure 2 show the fraction of patients achieving that degree of improvement for each study. The figures are cumulative, so that patients whose change from baseline is, for example, 50%, are also included at every level of improvement below 50%. Patients who did not complete the study were assigned 0% improvement. Some patients experienced a decrease in pain as early as Week 1, which persisted throughout the study.
For various degrees of improvement in pain from baseline to study endpoint, Figure 1 and Figure 2 show the fraction of patients achieving that degree of improvement for each study. The figures are cumulative, so that patients whose change from baseline is, for example, 50%, are also included at every level of improvement below 50%. Patients who did not complete the study were assigned 0% improvement. Some patients experienced a decrease in pain as early as Week 1, which persisted throughout the study.
 In an open label long-term uncontrolled study, pain reduction in patients responding to 8 weeks of acute treatment with duloxetine 60 mg once daily was maintained for a further 6 months as measured by change on the Brief Pain Inventory 24 hour average pain item. Patients who did not respond to 60 mg once daily in the acute phase or maintenance phase and were treated with duloxetine 120 mg once daily showed a decrease in pain intensity from baseline to endpoint.
In an open label long-term uncontrolled study, pain reduction in patients responding to 8 weeks of acute treatment with duloxetine 60 mg once daily was maintained for a further 6 months as measured by change on the Brief Pain Inventory 24 hour average pain item. Patients who did not respond to 60 mg once daily in the acute phase or maintenance phase and were treated with duloxetine 120 mg once daily showed a decrease in pain intensity from baseline to endpoint. Andepra at the recommended dose of 60 mg to 120 mg once daily demonstrated statistically significant superiority over placebo as measured by improvement in the Hamilton Anxiety Scale (HAM-A) total score and by the Sheehan Disability Scale (SDS) global functional impairment score.
Andepra at the recommended dose of 60 mg to 120 mg once daily demonstrated statistically significant superiority over placebo as measured by improvement in the Hamilton Anxiety Scale (HAM-A) total score and by the Sheehan Disability Scale (SDS) global functional impairment score. Duloxetine hydrochloride is a white to slightly brownish white solid, which is slightly soluble in water.
Duloxetine hydrochloride is a white to slightly brownish white solid, which is slightly soluble in water.