What is in this leaflet
This leaflet answers some common questions about Idacio.
It does not contain all the available information.
It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you using this medicine against the benefits they expect it will have for you.
If you have any concerns about using this medicine, ask your doctor or pharmacist.
Read this leaflet carefully before you use Idacio and keep it with the medicine. You may need to read it again.
What Idacio is used for
The active ingredient in this medicine is adalimumab, which is a fully human monoclonal antibody. Adalimumab recognises and binds to a specific protein (tumour necrosis factor or TNF-alpha), which is present at higher levels in some inflammatory diseases.
Idacio is intended for the treatment of a number of inflammatory diseases:
- Rheumatoid arthritis
Rheumatoid arthritis is an inflammatory disease of the joints. Signs and symptoms of rheumatoid arthritis include joint pain, tenderness, swelling and stiffness.
Idacio is used to reduce the signs and symptoms of moderate to severely active rheumatoid arthritis, as well as to slow down and protect the joints from further damage to help them move more freely.
Your doctor will decide if Idacio should be used with another medicine called methotrexate, or on its own.
Idacio can also be used to treat severe, active and progressive rheumatoid arthritis without previous methotrexate treatment. - Polyarticular Juvenile Idiopathic Arthritis (pJIA)
pJIA is an inflammatory disease of the joints. Idacio is used to reduce the signs and symptoms of moderate to severely active pJIA, in patients 2 years of age and older, when other medicines are not appropriate.
Your doctor will decide whether Idacio should be used with another medicine called methotrexate or used alone. - Enthesitis-related arthritis (ERA)
ERA is an inflammatory disease of the joints and the places where tendons join the bone. Idacio is used to treat ERA in children.
You may have already been given other medicines to treat your condition. Your doctor has prescribed Idacio for you as you haven't responded well enough to these medicines. - Psoriatic arthritis (PsA)
PsA is an inflammatory disease of the joints that is usually associated with psoriasis. Signs and symptoms include joint pain, tenderness and swelling. Idacio is used to reduce the signs and symptoms, of moderate to severely active PsA, as well as to slow down and protect the joints from further damage, to help them move more freely.
You may have already been given other medicines to treat your condition. Your doctor has prescribed Idacio for you as you haven't responded well enough to these medicines. - Ankylosing spondylitis
Ankylosing spondylitis is an inflammatory disease of the spine. Signs and symptoms of ankylosing spondylitis include back pain and stiffness. Idacio is used to reduce the signs and symptoms in patients with active disease. - Crohn's Disease
Crohn's disease is an inflammatory disease of the digestive tract. Idacio is used to treat moderate to severe Crohn's disease, in adults and children aged 6 years and over, to reduce the signs and symptoms of the disease and to induce and maintain periods where the symptoms are no longer present (remission).
You may have already been given other medicines to treat your condition. Your doctor has prescribed Idacio for you as you may either have not responded well enough, or you may have lost response or cannot tolerate these medicines. - Ulcerative Colitis
Ulcerative colitis is an inflammatory disease of the large intestine (bowel). Idacio is used to treat moderate to severe ulcerative colitis when other medicines are not appropriate. - Psoriasis
Psoriasis is an inflammatory disease of the skin. Plaque psoriasis, the most common form, is a skin condition that causes red, flaky, crusty patches of the skin covered with silvery scales. Plaque psoriasis can also affect nails, causing them to crumble, thicken and lift away from the nail bed which can be painful. Idacio is used to treat moderate to severe forms of the disease in adults and severe forms in adolescents and children from 4 years of age for whom topical therapy (such as creams, lotions and ointments) and phototherapy (also known as light therapy) have either not worked very well or are not suitable. - Hidradenitis suppurativa (HS)
HS (sometimes called acne inversa) is a chronic and often painful inflammatory skin disease. Symptoms may include tender nodules (lumps) and abscesses (boils) that may leak pus, which can have an unpleasant odour. It most commonly affects specific areas of the skin, such as under the breasts, the armpits, inner thighs, groin and buttocks. Scarring may also occur in affected areas.
Idacio is used for the treatment of adult and adolescents from 12 years of age with active moderate to severe HS. Idacio can reduce the number of nodules and abscesses caused by the disease, and the pain that is often associated with it.
You may have already been given other medicines to treat your condition. Your doctor has prescribed Idacio for you as you haven't responded well enough to these medicines.
Your doctor will schedule follow-up appointments to check on your progress to determine whether you should continue treatment. - Uveitis
Uveitis is an inflammatory disease affecting certain parts of the eye. This inflammation may lead to a decrease of vision and/or the presence of floaters in the eye (black dots or wispy lines that move across the field of vision). Idacio is used to treat non-infectious intermediate, posterior and pan-uveitis.
You may have already been given other medicines to treat your condition. Your doctor has prescribed Idacio for you as you may either have not responded well enough, or you have lost response or cannot tolerate these medicines.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is not addictive.
This medicine is only available with a doctor's prescription.
The long-term effects of Idacio on the growth and development of children is not known.
Before you use Idacio
When you must not use it
Do not use Idacio if you have an allergy to any medicine containing adalimumab or any of the ingredients listed at the end of this leaflet.
Symptoms of an allergic reaction may include:
- chest tightness
- shortness of breath, wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- hives, itching or skin rash.
Do not use Idacio if you have a severe infection including an infection of the blood (sepsis), active tuberculosis or other severe infections that can be caused by viruses, fungi, parasites or bacteria.
Infections can occur when the body's natural defences are lowered.
Do not use Idacio if you are already using anakinra (Kineret). Anakinra is a medicine for rheumatoid arthritis, JIA and conditions associated with a defect in a protein called cryoprin.
Do not use Idacio if you have moderate to severe heart failure.
If you are not sure whether any of the above conditions apply to you, ask your doctor.
Do not use this medicine after the expiry date printed on the label / blister / carton or if the packaging is torn or shows signs of tampering.
Return out of date or damaged medicines to your pharmacist for disposal.
Before you use it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if:
- you have or have had an infection, including a long-term infection or an infection in one part of the body (for example, leg ulcer).
- you have had infections which keep coming back or other conditions that increase the risk of infections.
If you are over 65, you may be more likely to get an infection while taking Idacio. It is important that you and your doctor pay special attention to signs of infection while you are being treated with Idacio. - you have ever had tuberculosis, or if you have been in close contact with someone who has had tuberculosis.
As cases of tuberculosis have been reported in patients treated with Idacio, your doctor will check you for signs and symptoms of tuberculosis before starting Idacio. This will include a thorough medical evaluation, including your medical history, and appropriate screening tests (for example, a chest x-ray and tuberculin test).
Tuberculosis can develop during therapy even if you have received treatment for the prevention of tuberculosis.
If symptoms of tuberculosis (for example a cough that doesn't go away, weight loss, lack of energy, mild fever), or any other infections appear during or after therapy, tell your doctor immediately. - you are a carrier of the hepatitis B virus (HBV), or you have active HBV or you think you might be at risk of contracting HBV.
In people who carry HBV, Idacio can cause the virus to become active again. In some rare cases, especially if you are taking other medicines that suppress the immune system, reactivation of HBV can be life threatening. - you have or have had a fungal infection or have lived or travelled in countries where some fungal infections are common. These infections may develop or become more severe if you take Idacio.
- you have or have had uveitis, your doctor may check for signs and symptoms of neurologic disease before starting this medicine.
- you have or develop a demyelinating disease (a disease that affects the insulating layer around the nerves, such as multiple sclerosis).
- you have or have had allergic reactions such as chest tightness, wheezing, dizziness, swelling or rash.
- you have or have had a blood disorder.
- you have or have had low resistance to disease.
- you have or have had a serious heart condition.
- you have or have had cancer or autoimmune disease.
- you have a lung disease called chronic obstructive pulmonary disease (COPD).
- you have or have had kidney or liver problems.
Tell your doctor if you are scheduled for any vaccines. Certain vaccines may cause infections and should not be given while patients are receiving Idacio.
Wherever possible, it is recommended that children be brought up to date with all immunisations according to current immunisation guidelines prior to starting Idacio therapy. Patients receiving Idacio should not receive live vaccines.
Tell your doctor if you have psoriasis and have undergone phototherapy, also known as light therapy.
Tell your doctor if you are pregnant or plan to become pregnant. You should consider the use of adequate contraception to prevent pregnancy and continue its use for at least 5 months after the last Idacio treatment.
Idacio should only be used during pregnancy if clearly needed.
A pregnancy study found that there was no higher risk of birth defects when the mother had used Idacio during pregnancy, compared with mothers with the same disease who did not use Idacio.
If you use Idacio during pregnancy, your baby may have a higher risk of getting an infection.
It is important that you tell your baby's doctors and other healthcare professionals about your Idacio use during your pregnancy before the baby receives any vaccine.
Tell your doctor if you are breastfeeding or plan to breastfeed. It is not known whether Idacio passes into breast milk. If you are breastfeeding, your doctor may advise you to stop breastfeeding while you are using this medicine.
If you have not told your doctor or pharmacist about any of the above, tell them before you start using Idacio.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you buy without a prescription from your pharmacy, supermarket or health food shop. Some medicines and Idacio may interfere with each other.
Tell your doctor or pharmacist if you are taking anakinra (Kineret) or abatacept (Orencia) Taking either of these two medicines together with Idacio may increase the risk of infection.
Tell your doctor if you are taking azathioprine or 6- mercaptopurine with Idacio.
Tell your doctor if you are taking any other medicines to treat your condition.
Idacio can be taken together with other medicines such as: methotrexate and other disease-modifying anti-rheumatic agents (for example, sulfasalazine, hydroxychloroquine, leflunomide and injectable gold preparations), steroids or pain medications including non-steroidal anti-inflammatory drugs (NSAIDS) such as ibuprofen.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while using this medicine.
How to use Idacio
Idacio is given as a subcutaneous injection (under the skin). It may be injected by the patient, family member or carer.
Follow all directions given to you by your doctor and pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the label or in this leaflet, ask your doctor or pharmacist for help.
Always use Idacio exactly as your doctor has instructed you.
You should check with your doctor or pharmacist if you are not sure.
Patients requiring a dose less than 40 mg should use the 40 mg vial presentation of Idacio.
How much to use
Rheumatoid Arthritis in Adults
The usual dose for adults with rheumatoid arthritis, is one 40mg injection every fortnight.
If you are receiving Idacio without methotrexate, your doctor may change your Idacio dose to 40 mg every week or 80 mg every fortnight, depending on your response.
Other medicines may be prescribed by your doctor to be taken while you are being treated with Idacio.
Psoriatic Arthritis & Ankylosing Spondylitis in Adults
The usual dose for patients with psoriatic arthritis and ankylosing spondylitis is one 40 mg injection every fortnight.
Other medicines may be prescribed by your doctor to be taken while you are being treated with Idacio.
Crohn's disease & Ulcerative Colitis in Adults
The usual dose for adults with Crohn's disease or ulcerative colitis is as follows:
- initial dose of 160 mg (day 1) (given as four 40 mg injections in one day OR as two 40 mg injections per day over two consecutive days) - 80 mg at day 15 (given as two 40 mg injections in one day)
- 40 mg starting at day 29, which then continues once every fortnight (maintenance dose).
Your doctor may change this ongoing (maintenance) dose to 40 mg every week or 80 mg every fortnight, depending on your response.
Psoriasis in Adults
The usual dose for adult patients with psoriasis is as follows:
- initial dose of 80 mg (day 1) (given as two 40 mg injections in one day)
- 40 mg given one week later (day 8), then
- 40 mg every fortnight from day 22 (maintenance dose).
Your doctor may change this ongoing (maintenance) dose to 40 mg every week or 80 mg every fortnight, depending on your response.
Uveitis in Adults
The usual dose for adults with uveitis is as follows:
- initial dose of 80 mg (day 1) (given as two 40 mg injections in one day),
- 40 mg one week later (day 8), then
- 40 mg every fortnight, starting at day 22, which then continues (maintenance dose).
Hidradenitis suppurativa in Adults
The usual dose for adults with HS is as follows:
- initial dose of 160 mg (day 1) (given as as four 40 mg injections in one day OR as two 40 mg injections per day over two consecutive days)
- 80 mg dose (day 15) (given as two 40 mg injections in one day)
- 40 mg every week or 80 mg every fortnight starting at day 29, which then continues (maintenance dose).
Your doctor may prescribe other medicines for your condition to take with Idacio.
Juvenile Idiopathic Arthritis & Enthesitis-Related Arthritis
The usual dose for children with pJIA, or ERA depends on body weight.
For a body weight of 30 kg and above:
The usual dose is 40 mg given every fortnight.
For a body weight between 10 kg and less than 30 kg:
The usual dose is 20 mg given every fortnight.
Crohn's disease in Children
The usual dose for children with Crohn's disease depends on body weight.
For a body weight of 40 kg or above:
- initial dose of 160 mg (day 1) (given as four 40 mg injections in one day OR as two 40 mg injections per day over two consecutive days)
- 80 mg two weeks later (day 15) (given as as two 40 mg injections in one day).
- 40 mg every fortnight starting at day 29, which then continues (maintenance dose).
Your doctor may change this ongoing (maintenance) dose to 40 mg every week or 80 mg every fortnight, depending on your response.
For a body weight of less than 40 kg:
- initial dose of 80 mg (day 1) (given as two 40 mg injections in one day)
- 40mg two weeks later (day 15)
- 20mg every fortnight, starting at day 29, which then continues (maintenance dose).
Your doctor may change this ongoing (maintenance) dose to 20 mg every week, depending on your response.
Treatment of Crohn's disease in children should be supported by good nutrition to allow appropriate growth.
Psoriasis in Children
The usual dose for children with psoriasis depends on body weight.
For a body weight of 40 kg and above:
The usual dose is 40mg given once every week for the first two weeks, then once every fortnight.
For a body weight of less than 40 kg:
The usual dose is 20 mg given once every week for the first two weeks, then once every fortnight.
Hidradenitis suppurativa in Adolescents
The usual dose for with HS, (from 12 years of age, weighing at least 30 kg) is as follows:
- initial dose of 80 mg (day 1) given as two 40 mg injections in one day),
- 40 mg one week later (day 8).
- 40 mg every fortnight starting at day 22, which then continues (maintenance dose).
Your doctor may change this ongoing (maintenance) dose to 40 mg every week or 80 mg every fortnight, depending on your response.
It is recommended you use an antiseptic wash daily on the affected areas.
How to use it
Idacio is injected under the skin. The injection can be self-administered or given by another person, for example a family member friend or carer, but only after proper training in injection technique.
If you are using the Idacio pen, instructions for preparing and giving an injection of Idacio are provided in the Injecting instructions supplied with the product.
Read these instructions carefully and follow them step by step. These instructions explain how to self-inject this medicine.
Do not attempt to self-inject until you are sure that you understand how to prepare and give the injection. Your doctor or his/her assistant will also show you best how to self-inject.
Keep Idacio out of the sight and reach of children.
If you are using the Idacio prefilled pen

Read carefully these entire instructions before using your Idacio prefilled pen.
Important information:
- Only use Idacio pre-filled pen if your healthcare professional has trained you how to use it correctly.
- The Idacio pre-filled pen comes as a ready to use single-use pre-filled pen to give a full dose of adalimumab.
- Always inject using the technique your healthcare professional taught you.
- Children under 12 years of age are not allowed to inject themselves and injection must be done by a trained adult.
- Keep the Idacio pre-filled pen out of the reach of children.
- Do not insert your fingers into the opening of the safety guard.
- Do not use an Idacio e-filled pen that has been frozen or left in direct sunlight.
Talk to your healthcare professional if you have any questions or concerns.
Get Familiar with your Idacio Pre-filled pen:
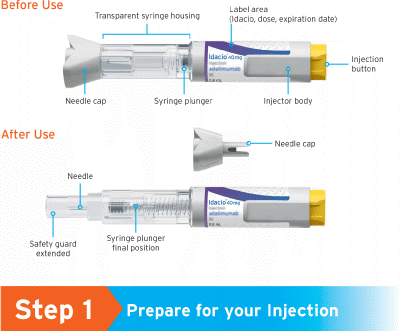
Each box of Idacio pre-filled pen comes with two pre-filled pens.
1.1 Prepare a clean flat surface, such as a table or countertop, in a well-lit area.
1.2 You will also need (Figure A):
- an alcohol pad (included in the box)
- a cotton ball or gauze, and
- a sharps disposal container.

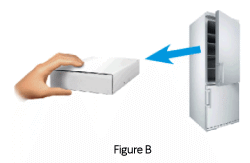
1.3 Remove the box from the refrigerator (Figure B).
1.4 Check the expiration date on the side of the box (Figure C).
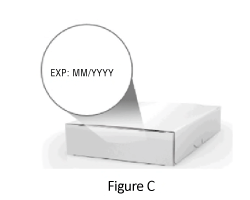
Warning: Do not use if expiration date has passed.
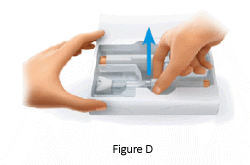
1.5 Take a pre-filled pen out of the original box:
- place two fingers on the label area
- pull the pre-filled pen straight up and out of the packaging (Figure D).
Put it on a clean flat surface.
1.6 Place the remaining pre-filled pen in its original box back in the refrigerator (Figure E).
Refer to Storage Information for how to store your unused pre-filled pen.
1.7 Leave the pre-filled pen at room temperature for at least 30 minutes to allow the medicine to warm up (Figure F).

Injecting cold medicine may be painful.
Warning: Do not warm the pre-filled pen any other way, such as in a microwave, hot water, or direct sunlight.
Warning: Do not remove the needle cap until you are ready to inject.

2.1 Wash your hands with soap and water (Figure G) and dry them well.
Warning: Gloves do not replace the need for washing hands.

3.1 Check the transparent syringe housing to make sure that:
- The liquid is clear, colorless, and free of particles (Figure H).
- The glass syringe is not cracked or broken (Figure H).
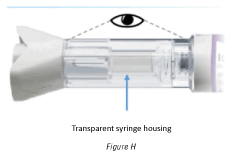
Warning: Do not use the pre-filled pen if the liquid contains particles, or is cloudy or if it is colored, or has flakes in it or shows any sign of damage.
If so, throw it away in a sharps disposal container and contact your healthcare professional or pharmacist.
3.2 Check the label to make sure that:
The name on the pre-filled pen says Idacio (Figure I).
The expiration date on the pre-filled pen has not passed (Figure I).
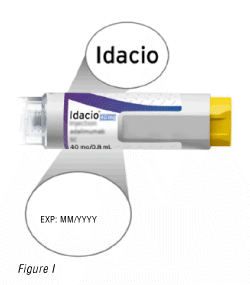
Warning: Do not use the pre-filled pen if the name on the label is not Idacio and/or the expiration date on the label has passed.
If so, throw away the pre-filled pen in a sharps disposal container and contact your healthcare professional or pharmacist.

4.1 Choose an injection site (Figure J) on:
- Top of the thighs.
- Abdomen (inject at least 5 centimeters away from the belly button).
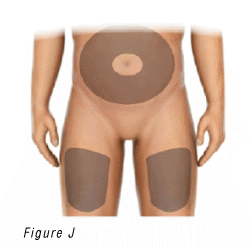
4.2 Choose a different site (at least 2.5 centimeters away from the previous injection site) each time to reduce redness, irritation or other skin problems.
Warning: Do not inject into an area that is sore (tender), bruised, red, hard, scarred or where you have stretch marks.
Warning: If you have psoriasis, do not inject into any lesions or red, thick, raised or scaly patches.

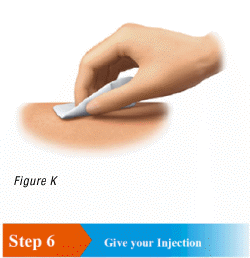
5.1 Wipe the skin of your injection site with an alcohol pad to clean it (Figure K).
Warning: Do not blow on or touch the injection site after cleaning.
6.1 Remove the needle cap
- Hold the pre-filled pen upwards and pull the needle cap straight off (Figure L).
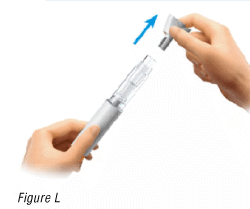
You may see drops of liquid at the needle tip.
- Throw away the needle cap.
Warning: Do not twist the cap.
Warning: Do not recap the pre-filled pen.
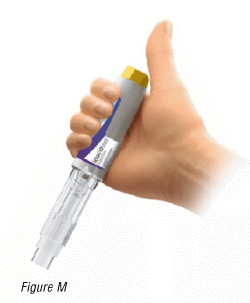
6.2 Position the pre-filled pen
- Hold the pre-filled pen so that you can see the transparent syringe housing.
- Place your thumb above (not touching) the yellow injection button (Figure M).
- Place the pre-filled pen against your skin at a 90° angle (Figure N).
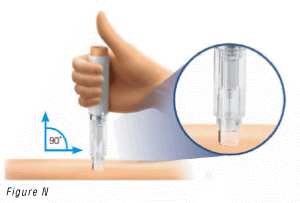
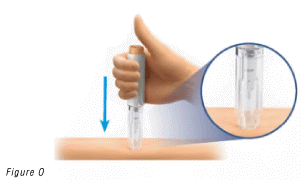
- Push and hold the pre-filled pen firmly against your skin until the safety guard is fully depressed.
This will unlock the injection button (Figure O).
6.3 Administer the Injection
- Push the injection button (Figure P).
You will hear a loud click, which means the injection has started.
- Continue to HOLD the pre-filled pen firmly.
- WATCH the syringe plunger to make sure it moves all the way down to the bottom (Figure P).
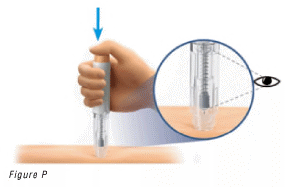
Warning: Do not lift the pre-filled pen from the skin until the plunger has moved all the way down and all the liquid has been injected.
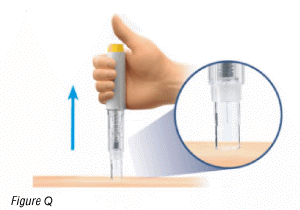
- When the syringe plunger has moved to the bottom and has stopped moving, continue holding it for 5 seconds.
- Lift the pre-filled pen from your skin (Figure Q).
The safety guard will slide down and lock into place to protect you from the needle (Figure Q).
Warning: Call your healthcare professional or pharmacist if you have any problem.
6.4 If there is blood or liquid on the skin, treat the injection site by gently pressing a cotton ball or gauze on the site (Figure R).
7.1 Throw away your used pre-filled pen in a sharps disposal container right away after use (Figure S).

Warning: Keep your sharps disposal container out of the reach of children.
Warning: Do not throw away the pre-filled pen in your household trash.
If you do not have a sharps disposal container, you may use a household container that is:
- Made of a heavy-duty plastic;
- Can be closed with a tight-fitting, puncture-resistant lid; that will keep sharps from coming out,
- Upright and stable during use,
- Leak-resistant and
- Properly labeled to warn of hazardous waste inside the container.
7.2 When your sharps disposal container is almost full, you will need to follow your local guidelines for the right way to dispose of your sharps disposal container.
Do not recycle your used sharps disposal container.

8.1 To help you remember when and where to do your next injection, you should keep a record of the dates and injection sites used for your injections (Figure T).

How long to use it
Keep using Idacio for as long as your doctor tells you.
Idacio will not cure your condition but should help your symptoms.
Ask your doctor if you are not sure how long to take the medicine for.
If you forget to use it
If you forget to give yourself an injection, you should inject the next dose of Idacio as soon as you remember. Then inject your next dose as you would have on your originally scheduled day.
Do not try to make up for missed doses by injecting more than one dose at a time.
If you use too much (overdose)
If you accidentally inject Idacio more frequently than prescribed by your doctor, immediately telephone your doctor or the Poisons Information Centre (Australia Telephone 13 11 26) or go to Accident and Emergency at your nearest hospital. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention. Always take the outer carton of the medicine with you.
While you are using Idacio
Things you must do
Check with your doctor before you receive any vaccines.
Wherever possible, it is recommended that children be brought up to date with all immunisations in according to current immunisation guidelines prior to starting Idacio therapy.
Patients receiving Idacio should not receive live vaccines (for example, BCG or oral polio vaccine).
If you become pregnant while using Idacio, tell your doctor immediately.
If you are about to be started on any new medicine, tell your doctor you are using Idacio.
Tell all doctors, dentists, and pharmacists who are treating you that you are using Idacio.
If you are going to have surgery, tell all doctors that you are using Idacio. Your doctor may recommend you discontinue using Idacio temporarily.
Keep all of your doctor's appointments so that your progress can be checked.
Things you must not do
Do not give Idacio to anyone else, even if they have the same condition as you.
Do not use Idacio to treat any other complaints unless your doctor tells you to.
Do not stop taking Idacio, without checking with your doctor.
Do not take Idacio and anakinra (Kineret) or Idacio and abatacept (Orencia) together. Taking either of these two medicines with Idacio may lead to an increased risk of developing a serious infection.
Things to be careful of
It is important to tell your doctor if you get symptoms of an infection, such as a fever, skin sores, feeling tired or any problems with your teeth and gums.
You might get infections more easily while you are receiving Idacio treatment. These infections may be serious and include tuberculosis, infections caused by viruses, fungi, parasites or bacteria, or other infections. Sepsis, an infection of the blood, may, in rare cases, be life-threatening.
Your doctor may recommend you discontinue Idacio if you develop an infection.
Be careful driving or operating machinery until you know how Idacio affects you.
The effects on your ability to drive and use machines whilst taking this medicine are not known.
Side Effects
Tell your doctor as soon as possible if you do not feel well while using Idacio or you have any problems using it.
Do this even if you do not think the problems are connected with the medicine or are not listed in this leaflet.
All medicines can have unwanted side effects. Sometimes they are serious, but most of the time they are not. You may need medical attention if you get some side-effects.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
Ask your doctor or pharmacist any questions you may have.
Tell your doctor immediately or go to Accident and Emergency at your nearest hospital if you experience any of the following:
- Signs of an allergic reaction such as:
- Chest tightness
- Shortness of breath, wheezing or difficulty breathing
- Swelling of the face, lips, tongue or other parts of the body
- Hives, itching or skin rash. - Signs and symptoms suggestive of heart failure such as shortness of breath with exertion or upon lying down or swelling of the feet
- Signs and symptoms suggestive of a blood disorder such as persistent fever, bruising, bleeding very easily, paleness.
The above list includes very serious side effects. You may need urgent medical attention or hospitalisation.
Tell your doctor as soon as possible if you notice any of the following:
- Signs of tuberculosis such as persistent cough, weight loss, listlessness, fever
- Signs of infection such as fever, lack of energy, skin sores, problems with your teeth or gums, burning when you pass urine
You might get infections more easily while you are receiving Idacio treatment. - Signs of nervous system disorders such as numbness or tingling throughout your body, arm or leg weakness, double vision
- Signs of soft tissue infection, such as a bump or open sore that doesn't heal.
The above list includes serious side effects. You may need urgent medical attention. Serious side effects are rare.
Tell your doctor if you notice any of the following and they worry you:
- Pain, swelling, redness or itching at the site of injection
- Cold, runny nose, sinus infection, sore throat, cough, congestion on the chest, asthma or worsening of asthma symptoms
- Lower respiratory tract infections (such as bronchitis, pneumonia)
- Pain in the ear which could suggest an ear infection
- Pain or inflammation of the eye or eye lid or changes to your vision
- Mouth ulcers, pain or excessive bleeding from the gums
- Burning or pain when passing urine, or blood in the urine
- Skin bumps or sores that don't heal
- Headache or migraine, dizziness, vertigo
- Muscle weakness or numbness, difficulty balancing
- Fever, flushing, increased sweating
- Nausea, vomiting, abdominal pain
- Reflux or heartburn
- Chest pain
- Rash, itching, redness or scaly patches
- Problems with your finger or toe nails
- Hair loss
- Fatigue, tiredness, lack of energy
- Muscle, joint or bone pain
- Bleeding or bruising more easily than usual
- Feeling overwhelmed or sad, or lacking motivation (depression)
- Feeling anxious, especially fearful or worried (anxiety)
- Increased heart rate
- Viral infections (including the flu, cold sore blisters, chicken pox and shingles)
- Bacterial infections (including urinary tract infection)
- Fungal Infections.
The above list includes the more common side effects of Idacio. They are usually mild and short-lived.
There have been cases of certain kinds of cancer in patients using Idacio or similar medicines. People with more serious rheumatoid arthritis that have had the disease for a long time may have a higher chance of getting a kind of cancer that affects the lymph system, called lymphoma, or that affects the blood, called leukaemia. If you take Idacio your risk may increase.
On rare occasions, a specific and severe type of lymphoma has been observed in patients taking Idacio.
Tell your doctor if new skin lesions (skin spots or sores) appear, or if existing lesions change appearance during or after Idacio treatment.
Very rare cases of skin cancer have been observed in patients taking Idacio.
If you have COPD, or are a heavy smoker, you should discuss with your doctor whether treatment with a TNF blocker is right for you. There have been cases of cancers other than lymphoma in patients with a specific type of lung disease called Chronic Obstructive Pulmonary Disease (COPD) treated with another TNF blocker.
Laboratory results
Some side effects observed with Idacio may not have symptoms and may only be discovered through blood tests. These include, most commonly, increased lipids, elevated liver enzymes and low levels of white blood cells, and red blood cells in the blood.
Tell your doctor or pharmacist if you notice anything else that is making you feel unwell. Other side effects not listed above may occur in some people.
After using Idacio
Keep your pre-filled pen in the pack until it is time to use it.
Keep Idacio in a refrigerator (2°C to 8°C). Do not freeze.
Keep Idacio in the refrigerator in away children cannot get to it.
Keep the medicine at the right temperature when you travel. This is important when travelling by car, bus, train, plane or any other form of transport.
Store a pre-filled pen at room temperature (below 25°C) for a maximum period of 14 days, protected from light.
Once removed from the refrigerator and stored at room temperature, the prefilled-pen must be used within 14 days or discarded, even if it is returned to the refrigerator.
Write down the date you first remove the pre-filled pen from the refrigerator on the label, so you can check how long it has been.
Disposal
After injecting Idacio, immediately throw away the used pre-filled pen in a special 'sharps' container as instructed by your doctor, nurse or pharmacist. If your doctor tells you to stop using Idacio or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product Description
What it looks like
Idacio is a clear, colourless solution.
Ingredients
Adalimumab
Monobasic sodium phosphate dihydrate
Dibasic sodium phosphate dihydrate
Mannitol
Sodium chloride
Citric acid monohydrate
Sodium citrate dihydrate
Polysorbate 80
Sodium hydroxide (for pH adjustment)
Water for injections
Supplier
Idacio is supplied in Australia by:
Fresenius Kabi Australia Pty Limited
Level 2, 2 Woodland Way Mount Kuring-gai, NSW, 2080 Australia
Telephone: (02) 9391 5555
Idacio is supplied in New Zealand by:
Fresenius Kabi New Zealand Limited
60 Pavilion Drive
Airport Oaks, Auckland 2022
New Zealand
Freecall: 0800 144 892
Australian Registration Number
Aust R 320242
® = Registered Trademark
This leaflet was prepared on 16 June 2020.
Published by MIMS May 2021
 Available data suggest that a clinical response is usually achieved within 12 weeks of treatment. Continued therapy should be carefully reconsidered in a patient not responding within this time period.
Available data suggest that a clinical response is usually achieved within 12 weeks of treatment. Continued therapy should be carefully reconsidered in a patient not responding within this time period. Some patients who experience a decrease in their response may benefit from an increase in dosage to 40 mg Idacio every week, or 80 mg fortnightly.
Some patients who experience a decrease in their response may benefit from an increase in dosage to 40 mg Idacio every week, or 80 mg fortnightly.
 Some patients may benefit from increasing the frequency to weekly if a disease flare or an inadequate response is experienced during maintenance dosing:
Some patients may benefit from increasing the frequency to weekly if a disease flare or an inadequate response is experienced during maintenance dosing: During maintenance treatment, corticosteroids may be tapered in accordance with clinical practice guidelines.
During maintenance treatment, corticosteroids may be tapered in accordance with clinical practice guidelines. If retreatment with Idacio is indicated, the above guidance on dose and treatment duration should be followed.
If retreatment with Idacio is indicated, the above guidance on dose and treatment duration should be followed. Table 7 contains adverse drug reactions (ADRs), which in some cases represent groups of related Preferred Terms to represent a medical concept. The ADRs presented in the table were included based on criteria including statistical significance, doubling in rate in adalimumab treated patients compared to placebo treated patients, a rate greater than 1% for adalimumab treated patients and medical importance assessment.
Table 7 contains adverse drug reactions (ADRs), which in some cases represent groups of related Preferred Terms to represent a medical concept. The ADRs presented in the table were included based on criteria including statistical significance, doubling in rate in adalimumab treated patients compared to placebo treated patients, a rate greater than 1% for adalimumab treated patients and medical importance assessment.

 EC50 estimates ranging from 0.8 to 1.4 micrograms/mL were obtained through pharmacokinetic/ pharmacodynamic modelling of swollen joint count, tender joint count and ACR20 response from patients participating in Phase II and III trials.
EC50 estimates ranging from 0.8 to 1.4 micrograms/mL were obtained through pharmacokinetic/ pharmacodynamic modelling of swollen joint count, tender joint count and ACR20 response from patients participating in Phase II and III trials. Patients receiving adalimumab 40 mg every week in RA Study II also achieved statistically significant ACR 20, 50 and 70 response rates of 53.4%, 35.0% and 18.4%, respectively, at six months. See Figure 2.
Patients receiving adalimumab 40 mg every week in RA Study II also achieved statistically significant ACR 20, 50 and 70 response rates of 53.4%, 35.0% and 18.4%, respectively, at six months. See Figure 2. The results of components of the ACR response criteria RA Study III are shown in Table 11.
The results of components of the ACR response criteria RA Study III are shown in Table 11. In RA Study III, 84.7% of patients with ACR20 responses at Week 24 maintained the response at 52 weeks. Clinical responses were maintained for up to 5 years in the open-label portion of RA Study III. ACR responses observed at Week 52 were maintained or increased through 5 years of continuous treatment with 22% (115/534) of patients achieving major clinical response. A total of 372 (67.8%) subjects had no change in their methotrexate dose during the study, 141 (25.7%) subjects had a dose reduction and 36 (6.6%) subjects required a dose increase. A total of 149 (55.6%) subjects had no change in their corticosteroid dose during the study, 80 (29.9%) subjects had a dose reduction and 39 (14.6%) subjects required a dose increase. Figure 3 illustrate the durability of ACR20 responses to adalimumab in RA Studies III and II.
In RA Study III, 84.7% of patients with ACR20 responses at Week 24 maintained the response at 52 weeks. Clinical responses were maintained for up to 5 years in the open-label portion of RA Study III. ACR responses observed at Week 52 were maintained or increased through 5 years of continuous treatment with 22% (115/534) of patients achieving major clinical response. A total of 372 (67.8%) subjects had no change in their methotrexate dose during the study, 141 (25.7%) subjects had a dose reduction and 36 (6.6%) subjects required a dose increase. A total of 149 (55.6%) subjects had no change in their corticosteroid dose during the study, 80 (29.9%) subjects had a dose reduction and 39 (14.6%) subjects required a dose increase. Figure 3 illustrate the durability of ACR20 responses to adalimumab in RA Studies III and II.
 In the open-label extension for RA study V, ACR responses were maintained when followed for up to 10 years. However, no statistical hypothesis was tested in the OLE period. Of 542 patients who were randomised to adalimumab 40 mg fortnightly, 170 patients continued on adalimumab 40 mg fortnightly for 10 years. Among those, 154 patients (90.6%) had ACR20 responses; 127 patients (74.7%) had ACR50 responses and 102 patients (60.0%) had ACR70 responses.
In the open-label extension for RA study V, ACR responses were maintained when followed for up to 10 years. However, no statistical hypothesis was tested in the OLE period. Of 542 patients who were randomised to adalimumab 40 mg fortnightly, 170 patients continued on adalimumab 40 mg fortnightly for 10 years. Among those, 154 patients (90.6%) had ACR20 responses; 127 patients (74.7%) had ACR50 responses and 102 patients (60.0%) had ACR70 responses.
 In RA Study V, adalimumab-treated patients had a mean duration of rheumatoid arthritis of less than 9 months and had not previously received methotrexate. Structural joint damage was assessed radiographically and expressed as change in modified Total Sharp Score. The Week 52 results are shown in Table 15. A statistically significant difference for change in modified Total Sharp Score and the erosion score was observed at Week 52 and maintained at Week 104.
In RA Study V, adalimumab-treated patients had a mean duration of rheumatoid arthritis of less than 9 months and had not previously received methotrexate. Structural joint damage was assessed radiographically and expressed as change in modified Total Sharp Score. The Week 52 results are shown in Table 15. A statistically significant difference for change in modified Total Sharp Score and the erosion score was observed at Week 52 and maintained at Week 104.
 At Year 2, 94/207 (45.4%) of patients who originally entered the study achieved a -0.5 reduction in HAQ. 79.5% (115/195) of the patients who achieved a reduction in HAQ of -0.5 at the end of one year of adalimumab treatment maintained this response over 5 years of active treatment.
At Year 2, 94/207 (45.4%) of patients who originally entered the study achieved a -0.5 reduction in HAQ. 79.5% (115/195) of the patients who achieved a reduction in HAQ of -0.5 at the end of one year of adalimumab treatment maintained this response over 5 years of active treatment.

 In subjects treated with adalimumab with no radiographic progression from baseline to Week 48 (n = 102), 84% continued to show no radiographic progression through 144 weeks of treatment.
In subjects treated with adalimumab with no radiographic progression from baseline to Week 48 (n = 102), 84% continued to show no radiographic progression through 144 weeks of treatment.
 A low level of disease activity (defined as a value < 20 [on a scale of 0-100 mm] in each of the four ASAS response parameters) was achieved at 24 weeks in 22% of adalimumab-treated patients vs. 6% in placebo-treated patients (p < 0.001). See Table 21.
A low level of disease activity (defined as a value < 20 [on a scale of 0-100 mm] in each of the four ASAS response parameters) was achieved at 24 weeks in 22% of adalimumab-treated patients vs. 6% in placebo-treated patients (p < 0.001). See Table 21. Results of this study were similar to those seen in the second randomised trial (AS Study II or M03-606), a multicenter, double-blind, placebo-controlled study of 82 patients with ankylosing spondylitis. Patient Reported Outcomes were assessed in both ankylosing spondylitis studies using the generic health status questionnaire SF-36 and the disease specific Ankylosing Spondylitis Quality of Life Questionnaire (ASQoL). The adalimumab-treated patients had significantly greater improvement in SF-36 Physical Component Score (mean change: 6.93) compared to placebo-treated patients (mean change: 1.55; p < 0.001) at Week 12, which was maintained through Week 24.
Results of this study were similar to those seen in the second randomised trial (AS Study II or M03-606), a multicenter, double-blind, placebo-controlled study of 82 patients with ankylosing spondylitis. Patient Reported Outcomes were assessed in both ankylosing spondylitis studies using the generic health status questionnaire SF-36 and the disease specific Ankylosing Spondylitis Quality of Life Questionnaire (ASQoL). The adalimumab-treated patients had significantly greater improvement in SF-36 Physical Component Score (mean change: 6.93) compared to placebo-treated patients (mean change: 1.55; p < 0.001) at Week 12, which was maintained through Week 24.
 Clinical remission results presented in Table 24 remained relatively constant irrespective of previous TNF antagonist exposure.
Clinical remission results presented in Table 24 remained relatively constant irrespective of previous TNF antagonist exposure. 117/854 patients had draining fistulas both at screening and at baseline. For the assessment of fistula healing, the data for both doses of adalimumab used in the study were pooled. The proportion of subjects (ITT population) with fistula healing at Week 26 was statistically significantly greater in patients treated with adalimumab [21/70 (30.0%)] compared to placebo [6/47 (12.8%)]. Complete fistula healing was maintained through Week 56 in 23/70 (32.9%) and 6/47 (12.8%) patients (ITT population) in the adalimumab and placebo groups, respectively.
117/854 patients had draining fistulas both at screening and at baseline. For the assessment of fistula healing, the data for both doses of adalimumab used in the study were pooled. The proportion of subjects (ITT population) with fistula healing at Week 26 was statistically significantly greater in patients treated with adalimumab [21/70 (30.0%)] compared to placebo [6/47 (12.8%)]. Complete fistula healing was maintained through Week 56 in 23/70 (32.9%) and 6/47 (12.8%) patients (ITT population) in the adalimumab and placebo groups, respectively.


 Statistically significant increases (improvement) from Baseline to Week 26 and 52 in Body Mass Index and height velocity were observed for both treatment groups. Statistically and clinically significant improvements from Baseline were also observed in both treatment groups for quality of life parameters (including IMPACT III).
Statistically significant increases (improvement) from Baseline to Week 26 and 52 in Body Mass Index and height velocity were observed for both treatment groups. Statistically and clinically significant improvements from Baseline were also observed in both treatment groups for quality of life parameters (including IMPACT III). Adalimumab should be discontinued in patients who do not achieve a clinical response during the first 8 weeks of therapy because very few patients will achieve clinical remission with continuing treatment. In UC-1 and UC-2, of patients given adalimumab 160/80 mg at baseline who did not achieve a clinical response at Week 8, 5.2%, and 17.0% went on to be in remission and response, respectively at Week 52. See Table 29.
Adalimumab should be discontinued in patients who do not achieve a clinical response during the first 8 weeks of therapy because very few patients will achieve clinical remission with continuing treatment. In UC-1 and UC-2, of patients given adalimumab 160/80 mg at baseline who did not achieve a clinical response at Week 8, 5.2%, and 17.0% went on to be in remission and response, respectively at Week 52. See Table 29. Statistically significant reductions of both all-cause and UC-related rates of hospitalisation were observed in a pooled analysis of Studies UC I and II.
Statistically significant reductions of both all-cause and UC-related rates of hospitalisation were observed in a pooled analysis of Studies UC I and II.
 Two of the continuous treatment populations entering trial M03-658 were those from Period C of Study I and those from Study II.
Two of the continuous treatment populations entering trial M03-658 were those from Period C of Study I and those from Study II. Of those who continued to receive adalimumab treatment until Week 52, 65.0% achieved mNAPSI 75 response and 61.3% achieved PGA-F response.
Of those who continued to receive adalimumab treatment until Week 52, 65.0% achieved mNAPSI 75 response and 61.3% achieved PGA-F response. Patients who achieved PASI 75 and PGA clear or minimal were withdrawn from treatment for up to 36 weeks and monitored for loss of disease control (loss of PGA response). Patients were then re-treated with adalimumab 0.8 mg/kg fortnightly for an additional 16 weeks. Among patients who were responders to the initial 16 weeks of treatment but who relapsed upon withdrawal and were retreated, PASI 75 response of 78.9% (15 of 19 subjects) and PGA clear or minimal of 52.6% (10 of 19 subjects) was observed.
Patients who achieved PASI 75 and PGA clear or minimal were withdrawn from treatment for up to 36 weeks and monitored for loss of disease control (loss of PGA response). Patients were then re-treated with adalimumab 0.8 mg/kg fortnightly for an additional 16 weeks. Among patients who were responders to the initial 16 weeks of treatment but who relapsed upon withdrawal and were retreated, PASI 75 response of 78.9% (15 of 19 subjects) and PGA clear or minimal of 52.6% (10 of 19 subjects) was observed. There is a statistically significantly higher HiSCR rate at Week 36 in patients who continued to receive weekly adalimumab compared to those who stopped adalimumab at Week 12.
There is a statistically significantly higher HiSCR rate at Week 36 in patients who continued to receive weekly adalimumab compared to those who stopped adalimumab at Week 12.

 In both studies, all components of the primary endpoint contributed cumulatively to the overall difference between adalimumab and placebo groups (Table 36).
In both studies, all components of the primary endpoint contributed cumulatively to the overall difference between adalimumab and placebo groups (Table 36). Additionally, in Study UV I, statistically significant differences in favour of adalimumab versus placebo were observed for the secondary endpoints changes in AC cell grade, vitreous haze grade, and log MAR BCVA (mean change from best state prior to Week 6 to the final visit; P Values: 0.011, < 0.001 and 0.003, respectively).
Additionally, in Study UV I, statistically significant differences in favour of adalimumab versus placebo were observed for the secondary endpoints changes in AC cell grade, vitreous haze grade, and log MAR BCVA (mean change from best state prior to Week 6 to the final visit; P Values: 0.011, < 0.001 and 0.003, respectively). Equivalence results for Idacio were further confirmed by similarity of efficacy responses observed up to 1 year in the follow-up of the study.
Equivalence results for Idacio were further confirmed by similarity of efficacy responses observed up to 1 year in the follow-up of the study. Following the administration of 24 mg/m2 (up to a maximum of 40 mg) subcutaneously fortnightly to patients with enthesitis-related arthritis, the mean trough steady-state (values measured at Week 24) serum adalimumab concentrations were 8.8 ± 6.6 micrograms/mL for adalimumab without concomitant methotrexate and 11.8 ± 4.3 micrograms/mL with concomitant methotrexate. Based on a population pharmacokinetic (PK) modelling approach, simulated steady-state adalimumab serum trough concentrations for a weight-based dosing regimen (20 mg adalimumab fortnightly for body weight < 30 kg and 40 mg adalimumab fortnightly for body weight ≥ 30 kg) were comparable to the simulated trough concentrations for the body surface area-based regimen.
Following the administration of 24 mg/m2 (up to a maximum of 40 mg) subcutaneously fortnightly to patients with enthesitis-related arthritis, the mean trough steady-state (values measured at Week 24) serum adalimumab concentrations were 8.8 ± 6.6 micrograms/mL for adalimumab without concomitant methotrexate and 11.8 ± 4.3 micrograms/mL with concomitant methotrexate. Based on a population pharmacokinetic (PK) modelling approach, simulated steady-state adalimumab serum trough concentrations for a weight-based dosing regimen (20 mg adalimumab fortnightly for body weight < 30 kg and 40 mg adalimumab fortnightly for body weight ≥ 30 kg) were comparable to the simulated trough concentrations for the body surface area-based regimen.