What is in this leaflet
This leaflet answers some of the common questions people ask about IRESSA. It does not contain all the available information.
It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor will have weighed the risks of you taking IRESSA against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What IRESSA is for
IRESSA belongs to a group of medicines called antineoplastics. These medicines work by stopping cancer cells from growing and multiplying.
IRESSA is used to treat non-small cell lung cancer, which is one type of lung cancer.
Your doctor will have explained why you are being treated with IRESSA and told you what dose to take.
Your doctor may prescribe this medicine for another use. Ask your doctor if you want more information.
This medicine is available only with a doctor's prescription.
IRESSA is not addictive.
Before you use IRESSA
When you must not use it
You must tell your doctor if you have allergies to:
- gefitinib, the active ingredient in IRESSA
- any of the ingredients listed at the end of this leaflet.
Symptoms of an allergic reaction to IRESSA may include:
- shortness of breath, wheezing, difficulty breathing or a tight feeling in your chest
- swelling of the face, lips, tongue or other parts of the body
- rash, itching, hives or flushed, red skin
- dizziness or light-headedness
- back pain
Do not use IRESSA if you are pregnant. It is not known if it is safe for you to take IRESSA while you are pregnant. It may affect your baby if you take it at any time during pregnancy.
Do not breastfeed while taking IRESSA. IRESSA passes into breast milk and therefore there is a possibility that the breast-fed baby may be affected.
Do not give IRESSA to children. There is no experience of its use in children.
Do not take IRESSA after the use by (expiry) date printed on the pack.
Do not take IRESSA if the packaging is torn or shows signs of tampering.
Do not use it to treat any other complaints unless your doctor tells you to.
Do not give this medicine to anyone else.
Before you start to use it
You must tell your doctor if:
- you have any eye problems.
You may need further examination.
- you have any lung problems such as pneumonia
- you have any liver problems.
Taking other medicines
Tell your doctor if you are taking any other medicines, including:
- itraconazole & ketoconazole (medicines used to treat fungal infections)
- metoprolol (beta-blocker)
- rifampicin (antibiotic)
- warfarin (blood thinner)
- phenytoin, carbamazepine, barbiturates, St John's Wort
- vinorelbine (cancer drug)
- medicines that alter the acidity of the stomach e.g. antacids, H2 antagonists and proton pump inhibitors
- medicines that you buy at the chemist, supermarket or health food shop
These medicines may be affected by IRESSA, or may affect how well it works. You may need different amounts of your medicine, or you may need to take/use/have different medicines. Your doctor or pharmacist will advise you.
Your doctor and pharmacist may have more information on medicines to be careful with or avoid while taking IRESSA.
If you have not told your doctor about any of these things, tell them before you start taking any IRESSA.
Taking IRESSA
Follow all directions given to you by your doctor or pharmacist carefully.
They may differ from the information contained in this leaflet.
Your doctor or pharmacist will tell you how many tablets you will need to take each day.
If you do not understand the instructions on the box, ask your doctor or pharmacist for help.
How much to take
The usual adult dose is one 250 mg tablet taken each day.
Swallow your IRESSA tablet whole with a glass of water. Do not chew or crush the tablets.
For patients with swallowing difficulties the tablet can be dissolved in drinking water (non-carbonated). No other liquids should be used. Follow these instructions carefully:
- Drop the tablet into half a glass of drinking water (non-carbonated). DO NOT CRUSH THE TABLET.
- Stir the water until the tablet dissolves and the water becomes a cloudy pale orange solution (approximately 10 minutes).
- Drink the liquid immediately.
- Rinse the empty glass with half a glass of drinking water and drink the water.
This cloudy pale orange solution can also be given to patients via a nasogastric tube.
When to take it
Take IRESSA at about the same time each day. Taking IRESSA at the same time each day will help you remember to take it.
It does not matter if you take IRESSA with or without food.
How long to take it
Continue taking IRESSA for as long as your doctor tells you.
If you forget to take it
If you miss a dose take it as soon as you remember, as long as it is 12 hours before the next dose is due. If it is less than 12 hours to the next dose do not take the dose you have missed.
Do not take a double dose to make up for the dose that you missed.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
Overdose
Immediately telephone your doctor or pharmacist or the Poisons Information Centre (telephone 13 11 26), or go to Accident and Emergency at your nearest hospital, if you think that you or anyone else may have taken too much IRESSA. Do this even if there are no signs of discomfort or poisoning.
Symptoms of an IRESSA overdose include the side effects listed below in the 'Side Effects' section, but are usually of a more severe nature.
While you are using it
Things you must do
Be sure to keep all your appointments with your doctor so your progress can be checked. Your doctor may want to do some tests from time to time to check your progress and investigate any unwanted side effects.
If you become pregnant tell your doctor immediately.
Tell any other doctors, dentists and pharmacists who are treating you that you are taking IRESSA.
If you are about to be started on any new medicine, tell your doctor, dentist or pharmacist that you are taking IRESSA.
If you go into hospital, please let the medical staff know you are taking IRESSA.
Things you must not do
Do not give IRESSA to anyone else, even if they have the same condition as you.
Do not take IRESSA to treat any other complaints unless your doctor tells you to.
Do not stop taking IRESSA without checking with your doctor.
Things to be careful of
Be careful driving or operating machinery until you know how IRESSA affects you.
Some patients may feel weak.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking IRESSA.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical treatment if you get some of the side effects.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor as soon as possible if you notice any of the following and they worry you:
- skin reactions such as acne-like rash, sometimes itchy with dry skin
- nausea
- vomiting
- loss of appetite or weight changes
- red or sore mouth
- dry mouth
- problems with your nails
- hair loss
- unusual tiredness or weakness
- eye problems, including red and itchy eye, red and sore eyelid, dry eyes
- increased bleeding (e.g. nose bleeds, blood in your urine)
- fever
- burning sensations during urination and frequent, urgent need to urinate
- Skin reaction on the palms of the hands and soles of the feet including tingling, numbness, pain, swelling or reddening (known as palmar-plantar erythrodysaesthesia syndrome or hand and foot syndrome)
IRESSA may be associated with changes in your blood, urine or liver. Your doctor may want to perform tests from time to time to check on your progress and detect any unwanted side effects.
These are possible side effects of IRESSA.
Tell your doctor immediately if you notice any of the following:
- severe skin reactions with lesions, ulcers or blisters
- diarrhoea
- severe eye problems including an eye ulcer, sometimes with ingrowing eyelashes
- unexplained breathing problems
These side effects may be serious.
Tell your doctor immediately or go to Accident and Emergency at your nearest hospital if any of the following happen.
- rash, itching or hives on the skin,
- swelling of the face, lips, tongue or other parts of the body including hands, feet or ankles
- breathlessness/shortness of breath, wheezing or trouble breathing
- liver pain or swelling and/or a general feeling of unwell with or without jaundice (yellowing of the skin or eyes)
Some patients taking IRESSA get an inflammation of the lungs called interstitial lung disease. This side effect is common.
These are all serious side effects. You may need urgent medical attention or hospitalisation.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Some people may get other effects while taking IRESSA.
After using it
Storage
Keep your IRESSA tablets in the blister foil until it is time to take them. If you take IRESSA out of the blister foil, it will not keep well.
Keep it in a cool dry place where the temperature stays below 30°C.
Do not store it or any other medicine in the bathroom or near a sink.
Do not leave it on a window sill or in the car on hot days. Heat and dampness can destroy some medicines.
Keep your medicine where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
Ask your pharmacist what to do with any tablets you have left over if your doctor tells you to stop taking them, or you find that the expiry date has passed.
Product description
What IRESSA looks like
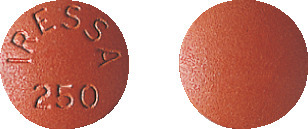
IRESSA tablets are brown, round, film-coated tablets. One side of the tablet is marked "IRESSA 250".
IRESSA tablets are packed in blister foils of 30 tablets.
Ingredients
Each IRESSA tablet contains 250 mg of gefitinib as the active ingredient.
Other ingredients:
- lactose monohydrate
- microcrystalline cellulose
- croscarmellose sodium
- povidone
- sodium lauryl sulphate
- magnesium stearate
- hypromellose
- macrogol 300
- titanium dioxide
- yellow iron oxide CI 77492
- red iron oxide CI 77491.
Supplier
IRESSA is sponsored and supplied in Australia by:
AstraZeneca Pty Ltd
ABN 54 009 805 342
66 Talavera Road
MACQUARIE PARK NSW 2113
Phone 1800 805342
This leaflet was prepared on January 2021
AUST R 90010
IRESSA 250 mg
Doc ID-003508982 v3.0
Published by MIMS March 2021

 Quality of life outcomes differed according to EGFR mutation status. In EGFR mutation-positive patients, significantly more Iressa-treated patients experienced an improvement in quality of life and lung cancer symptoms versus carboplatin/ paclitaxel whereas the reverse was the case in EGFR mutation negative patients (see Table 3).
Quality of life outcomes differed according to EGFR mutation status. In EGFR mutation-positive patients, significantly more Iressa-treated patients experienced an improvement in quality of life and lung cancer symptoms versus carboplatin/ paclitaxel whereas the reverse was the case in EGFR mutation negative patients (see Table 3).
 The co-primary analysis evaluating the overall survival in 174 patients with high EGFR gene copy number did not demonstrate superiority of Iressa over docetaxel. Survival outcomes in patients with high EGFR gene copy number were similar for both treatments (HR 1.09, 95% CI 0.78 to 1.51, p = 0.6199, median 8.4 vs 7.5 months).
The co-primary analysis evaluating the overall survival in 174 patients with high EGFR gene copy number did not demonstrate superiority of Iressa over docetaxel. Survival outcomes in patients with high EGFR gene copy number were similar for both treatments (HR 1.09, 95% CI 0.78 to 1.51, p = 0.6199, median 8.4 vs 7.5 months). Exploratory analysis of EGFR biomarkers was statistically inconclusive; however, response rates in ISEL were higher among Iressa treated patients with EGFR mutation-positive tumours.
Exploratory analysis of EGFR biomarkers was statistically inconclusive; however, response rates in ISEL were higher among Iressa treated patients with EGFR mutation-positive tumours. These data are consistent with the pre-planned exploratory Japanese subgroup analysis in IPASS. In that study ctDNA derived from serum, not plasma was used for EGFR mutation analysis using the EGFR Mutation Test Kit (DxS) (N = 86). In that study, concordance was 66%, sensitivity was 43.1%, specificity was 100%. The positive and negative predictive values were 100% and 54.7%, respectively.
These data are consistent with the pre-planned exploratory Japanese subgroup analysis in IPASS. In that study ctDNA derived from serum, not plasma was used for EGFR mutation analysis using the EGFR Mutation Test Kit (DxS) (N = 86). In that study, concordance was 66%, sensitivity was 43.1%, specificity was 100%. The positive and negative predictive values were 100% and 54.7%, respectively. Chemical name: N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy) quinazolin-4-amine.
Chemical name: N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy) quinazolin-4-amine.