What is in this leaflet
This leaflet answers some common questions about PROGRAF Capsules, ADVAGRAF XL Prolonged-Release Capsules and PROGRAF Concentrated Injection. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking PROGRAF or ADVAGRAF XL against the benefits this medicine is expected to have for you.
If you have any concerns about using PROGRAF or ADVAGRAF XL ask your doctor or pharmacist.
Keep this leaflet with your medicine. You may need to read it again.
What PROGRAF / ADVAGRAF XL is used for
You have been given a new transplanted liver, kidney, lung or heart from another person because your own was no longer healthy. Your body recognises that this new organ is different from your other organs and will try to reject it by attacking it in the same way that it would attack germs that enter your body. This could make you become ill again. PROGRAF or ADVAGRAF XL stops this attack; it is very important to take PROGRAF or ADVAGRAF XL given to you by your doctor regularly so that your new liver, kidney, lung or heart will not be attacked or rejected.
If you have been taking other medicines for this purpose, but are still feeling unwell, your doctor may change your treatment and begin giving you PROGRAF or ADVAGRAF XL.
PROGRAF or ADVAGRAF XL contains the active ingredient tacrolimus, which is an immunosuppressive agent.
Your doctor may have prescribed PROGRAF or ADVAGRAF XL for another reason.
Ask your doctor if you have any questions about why this medicine has been prescribed for you.
Before you use PROGRAF or ADVAGRAF XL
When you must not use it
Do not use PROGRAF or ADVAGRAF XL if you have an allergy to:
- any medicine containing tacrolimus or other macrolides (these are antibiotics of the erythromycin family - trade names are Eryc, EES, Klacid, Zithromax, Rulide and Biaxsig)
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Do not use PROGRAF or ADVAGRAF XL if the packaging is torn or shows signs of tampering. Do not use PROGRAF or ADVAGRAF XL beyond the expiry date printed on the pack.
Before you start to use it
You must tell your doctor if
- you are pregnant or planning to become pregnant
- you are using oral contraceptives
- you are breast feeding
- you are receiving cyclosporin immunosuppressive therapy
If you have not told your doctor or pharmacist about any of the above, tell them before you start taking or are given PROGRAF or ADVAGRAF XL.
Your doctor will advise you whether or not to take PROGRAF or ADVAGRAF XL or if you need to adjust the dose or adapt your treatment.
Taking other medicines:
Tell your doctor or pharmacist if you are taking any other medicines, including medicines you get without a prescription from a pharmacy, supermarket or health food shop.
Some medicines and PROGRAF or ADVAGRAF XL may interfere with each other.
Among these medicines is the herbal preparation St John's Wort (Hypericum perforatum) which is capable of decreasing tacrolimus blood levels.
Your doctor or pharmacist can tell you what to do if you are taking any of these medicines.
Effects on driving and operating machinery
- PROGRAF or ADVAGRAF XL may cause visual or nervous disturbances. If affected, do not drive or operate machinery.
Effects of food and alcohol
- Food reduces the absorption of PROGRAF or ADVAGRAF XL. Therefore, the capsules should be taken at least 1 hour before a meal.
Using PROGRAF or ADVAGRAF XL
How much to take:
You can only get PROGRAF or ADVAGRAF XL from your doctor. Your dose will be calculated according to your weight, age, and medical condition. As your health and the function of your new liver, kidney, lung or heart can be affected by how much medicine you take, it is normal that your doctor tests samples of your blood and urine at regular intervals. This is in order to test whether your medicine requires adjustment.
PROGRAF should be taken in two doses (e.g. morning and evening) each day. ADVAGRAF XL should be taken once a day in the morning. Take the capsule from the blister pack and swallow it whole with plenty of water. Do not use grapefruit juice as it contains substances that interfere with the action of PROGRAF or ADVAGRAF XL.
How to take it:
- PROGRAF or ADVAGRAF XL capsules should be taken at least 1 hour before a meal.
- If you receive PROGRAF Concentrated Injection by intravenous infusion, it will be added to a larger volume of diluent and infused over a prolonged period of time (usually 24 hours).
- You must never change the dose yourself even if you are feeling better. It is very important that you keep taking this medicine so that your body will not reject your new liver, kidney, lung or heart.
- If you accidentally take a larger dose than recommended, tell your doctor immediately.
- If you do not understand the instructions provided with this medicine, ask your doctor or pharmacist for help.
If you forget to take it
- If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
- Do not take a double dose to make up for the dose you missed.
If you have missed more than one dose, or are not sure what to do, check with your doctor or pharmacist.
If you have trouble remembering when to take your medicine, ask your pharmacist for some hints.
If you have taken too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at your nearest hospital.
Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
While you are taking PROGRAF or ADVAGRAF XL
Things you must do
- Always follow your doctor's instructions carefully
- Tell your doctor if you become pregnant while taking PROGRAF or ADVAGRAF XL
- If you are about to start taking a new medicine, tell your doctor and pharmacist that you are taking PROGRAF or ADVAGRAF XL.
- PROGRAF or ADVAGRAF XL suppresses your immune system by lowering your body's immune defence system. This increases your risk of skin cancer and other cancers while taking PROGRAF or ADVAGRAF XL. You should always protect yourself from the sun, wear sunscreen, a hat and protective clothing.
Things you must not do
- Do not take PROGRAF or ADVAGRAF XL to treat any other complaint unless your doctor says so.
- Do not give this medicine to anyone else, even if their symptoms are similar to yours.
Side Effects
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical treatment if you get some side effects.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor if you experience any of the following and they worry you:
- tiredness, lack of energy
- stomach upset, including nausea (feeling sick), vomiting, loss of appetite, diarrhoea, stomach cramps
- tremor (shaking)
- headache
- feeling depressed (sad)
- sleeping difficulties
- blurred vision or sensitive to light
- muscle cramps, tenderness or weakness
Tell your doctor immediately if you notice any of the following as you may need urgent medical care:
- signs of allergy such as rash, itching or hives on the skin; swelling of the face, lips, tongue or other part of the body; shortness of breath, wheezing or troubled breathing
- fever
- diabetes / increased blood sugar
- swelling, numbness or tingling (pins and needles) in your hands and feet
- constant "flu-like" symptoms such as chills, sore throat, aching joints, swollen glands, or any other signs of infection
- unusual bleeding or bruising
- high blood pressure
- palpitations, abnormal heart rhythms, chest pain
- new lumps or moles, or changes to existing moles, anywhere on the body
- swelling of the eyelids, hands or feet due to excess fluid
- a change in the amount of urine passed or in the number of times you urinate, pain on urinating, or other kidney problems.
- yellowing of the skin and/or eyes (jaundice) often accompanied by generally feeling unwell (for example, tiredness, lack of energy, loss of appetite, nausea and vomiting, pain in the abdomen)
- symptoms of anaemia, such as shortness of breath, tiredness or dizziness
- seizures (fits)
- buzzing or ringing in the ears, difficulty hearing
Other side effects not listed above may also occur in some people. Tell your doctor if you notice any other side effects.
After using PROGRAF or ADVAGRAF XL
Storage
Use all the capsules within 12 months of opening the aluminium wrapper.
Keep PROGRAF or ADVAGRAF XL Capsules in the blisters until it is time to take them.
Keep PROGRAF Capsules in a cool dry place where the temperature is below 30°C.
Keep ADVAGRAF XL Capsules in a cool dry place where the temperature is below 25°C.
Keep PROGRAF Concentrated Injection away from light and store below 25°C.
Keep your medicines where children cannot reach them. A locked cupboard at least one-and-a-half metres (1.5 m) above the ground is a good place to store medicines.
Do not store PROGRAF or ADVAGRAF XL, or any other medicine, in the bathroom or near a sink. Do not leave medicines in the car or on window sills. Heat and dampness can destroy some medicines.
Disposal
If your doctor tells you to stop taking PROGRAF or ADVAGRAF XL Capsules, or your medicine has passed its expiry date, ask your pharmacist what to do with any medicine which may be left over.
Product Description
What it looks like
PROGRAF
- PROGRAF 0.5 mg capsules (AUST R 77280) are light yellow with "0.5 mg" and "[f]607" printed in red, packed in blister sheets of ten capsules and sealed in an aluminium wrapper (Pack size 100 capsules).
- PROGRAF 1 mg capsules (AUST R 58930) are white with "1 mg" and "[f]617" printed in red, packed in blister sheets of ten capsules and sealed in an aluminium wrapper (Pack size 100 capsules).
- PROGRAF 5 mg capsules (AUST R 58931) are greyish-red with "5 mg" and "[f]657" printed in white (Pack size 50 capsules).
ADVAGRAF XL
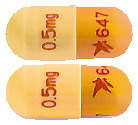
- ADVAGRAF XL 0.5mg prolonged-release capsules (AUST R 269212) are oblong capsules with a light yellow cap imprinted with "0.5 mg" and an orange body imprinted with "647" (Pack size 30 capsules).
- ADVAGRAF XL 1mg prolonged-release capsules (AUST R 269214) are oblong capsules with a white cap imprinted with "1 mg" and an orange body imprinted with "677" (Pack size 60 capsules).
- ADVAGRAF XL 3 mg prolonged-release capsules (AUST R 306501) are oblong capsules with an orange cap imprinted with "3 mg" and an orange body imprinted with "637" (Pack size: 50 capsules).
- ADVAGRAF XL 5mg prolonged-release capsules (AUST R 269215) are oblong capsules with a greyish red cap imprinted with "5 mg" and an orange body imprinted with "687" (Pack size 30 capsules).
PROGRAF Concentrated Injection
- PROGRAF Concentrated Injection ampoules (AUST R 58932) contain 5 mg tacrolimus in 1 mL (Pack size 10 ampoules).
Ingredients
PROGRAF Capsules contain hypromellose 2910, cellulose, croscarmellose sodium, lactose, and magnesium stearate. The capsule shell contains gelatin, purified water, titanium oxide and a dye (ferric oxide yellow E172 for 0.5 and 1 mg capsules and ferric oxide red E172 for 5 mg capsules. The 0.5 mg and 1mg capsules are printed with an ink containing shellac, soya lecithin, dimethylpolysiloxane and ferric oxide red E172, and the 5mg capsules with shellac, propylene glycol and titanium dioxide E171.
ADVAGRAF XL prolonged-release capsules contain hypromellose, ethylcellulose, lactose and magnesium stearate. The capsule shells contain gelatin, titanium dioxide, iron oxide yellow CI77492, iron oxide red CI77491 and sodium lauryl sulfate. The capsules also have a trace of printing ink, Opacode S-1-15083 red, which contains shellac, soya lecithin, simethicone and iron oxide red CI77491.
PROGRAF Concentrated Injection contains polyoxyethylene (PEG-60) hydrogenated castor oil and ethanol.
Sponsor
PROGRAF and ADVAGRAF XL are supplied in Australia by:
Astellas Pharma Australia Pty Ltd
6 Eden Park Drive
Macquarie Park NSW 2113
Tel: 1800 751 755 (Medical Information)
Email: aaumedinfo@astellas.com
(Medical Information)
Website: www.astellas.com/.au
® = Registered Trademark
This leaflet was prepared in January 2020.
Published by MIMS April 2020

